 |
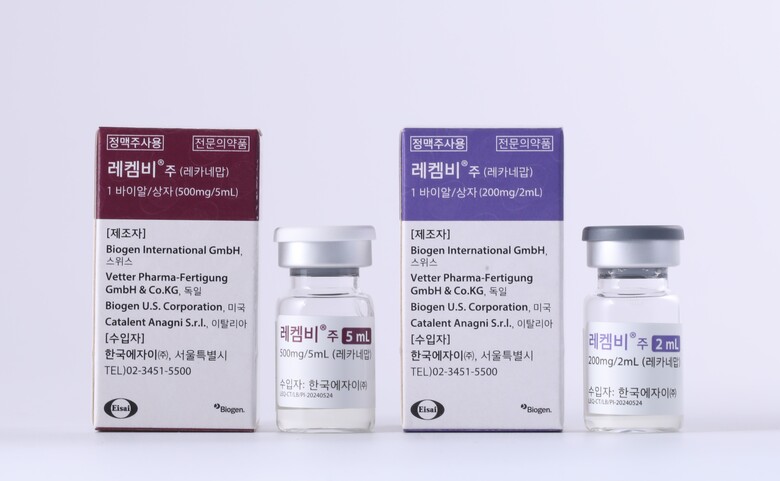
| ▲ 레켐비주(Leqembi, 성분명 레카네맙) |
[mdtoday=박성하 기자] 초기 알츠하이머병 치료제로 국내 허가를 받은 레카네맙(제품명 레켐비)을 둘러싸고 허가 과정의 적절성을 문제 삼는 목소리가 제기됐다. 시민단체 바른의료연구소는 해당 의약품이 충분한 근거 검증 없이 신속하게 승인됐다며 식품의약품안전처에 대한 철저한 감사를 촉구했다.
바른의료연구소는 성명을 통해 “레켐비는 한국인 대상 임상 근거에서 통계적 유의성이 명확히 확인되지 않았음에도 불구하고, 가교임상시험이 면제되고 중앙약사심의위원회 자문 절차마저 생략됐다”고 지적했다. 이는 규제 당국이 신약 허가 과정에서 요구되는 최소한의 안전성·유효성 검증을 충분히 수행하지 않았다는 문제 제기다.
레켐비는 지난해 5월 식약처의 품목 허가를 받은 뒤 같은 해 11월 국내 시장에 출시됐다. 현재 일부 상급종합병원과 대학병원을 중심으로 비급여 처방이 이뤄지고 있다.
문제의 핵심은 가교임상 면제 결정이다. 바른의료연구소에 따르면 레켐비의 글로벌 임상 3상에는 총 1795명이 참여했으며, 이 중 한국인은 130명에 불과하다. 식약처는 전체 임상에서 인지기능 평가지표(CDR-SB)가 위약 대비 27.1% 개선됐고, 한국인 하위그룹에서도 25.2%의 개선 효과가 나타났다는 점을 근거로 가교시험을 면제했다.
그러나 바른의료연구소는 “가교자료 평가 가이드라인은 다국가 임상 전체 결과와 한국인 하위 분석 결과를 통계적으로 비교·검증하도록 명시하고 있다”며 “레켐비 자료에는 전체 분석에 대한 p값은 제시됐지만, 한국인 하위 분석에서는 통계적 유의성이 구체적으로 제시되지 않았다”고 반박했다. 수치상 유사성이 곧 과학적 동등성을 의미하지는 않는다는 것이다.
국제 규제 흐름과의 비교도 문제로 지적됐다. 바른의료연구소는 “미국 식품의약국(FDA)이 레켐비를 가속 승인한 이후에도 임상적 유효성과 안전성을 둘러싼 논란은 계속되고 있으며, 유럽의약품청(EMA)은 보다 엄격한 기준을 적용해 허가 여부를 검토해 왔다”고 설명했다. 일부 국가에서는 비용 대비 효과성과 부작용 위험을 이유로 실제 임상 적용에 신중한 태도를 유지하고 있다는 점도 언급했다. 이러한 상황에서 한국 규제당국이 가교시험과 전문가 심의를 동시에 생략한 결정은 국제 규제 기조와도 괴리가 있다는 것이다.
중앙약사심의위원회 자문이 이뤄지지 않은 점도 도마에 올랐다. 중앙약심은 의약품의 안전성·유효성, 부작용 피해 구제 등을 심의하는 공식 자문기구다. 바른의료연구소는 “치매처럼 사회적 영향이 큰 질환의 치료제를 허가하면서 전문가 집단의 다층적 검증 절차를 생략한 것은 납득하기 어렵다”고 비판했다.
이어 “의무 규정이 없다는 이유로 핵심 심의 절차를 건너뛴 것이 단순 행정 편의인지, 특정 결론을 전제로 한 결정이었는지는 명확히 밝혀져야 한다”며 “만약 안전장치가 의도적으로 배제됐다면 이는 국민 신뢰를 훼손한 중대한 문제”라고 강조했다.
바른의료연구소는 특히 알츠하이머 환자와 가족들의 절박한 상황을 언급하며 신중한 접근을 촉구했다. 이 단체는 “근거가 충분하지 않은 치료제를 ‘희망’이라는 이름으로 포장해 유통하는 것은 윤리적으로도 문제가 있다”며 “식약처는 레켐비 허가 과정 전반을 투명하게 공개해야 한다”고 주장했다.
아울러 재발 방지를 위해 제도 개선도 요구했다. 고위험 신약의 경우 전문가 심의를 의무화하고, 이해충돌 방지 장치를 강화하는 등 허가 행정의 투명성을 높일 수 있는 법적 장치 마련이 필요하다는 입장이다. 이와 함께 레켐비 허가 과정에서 특혜나 절차상 문제 여부를 확인하기 위한 식약처 감사 실시를 정부에 촉구했다.
메디컬투데이 박성하 기자(applek99@mdtoday.co.kr)


[저작권자ⓒ 메디컬투데이. 무단전재-재배포 금지]